Google-Scholar Profile I am a biochemist by training with a mixed computational & experimental background. My early research focused on the biophysics of protein-protein interactions and structural dynamics using computational tools such as MD simulations, docking algorithms and lots of python programming. I later drifted back into the wet lab, helped developing standards and training programs for the emerging field of synthetic biology and started applying these principles to protein engineering. Ongoing collaborations with brilliant material scientists fuse our designer proteins with their designer materials and devices.
| Bio(electronic) Sensors |
| in collaboration with Sahika Inal and her Organic Biolectronic Laboratory |
|
|
|
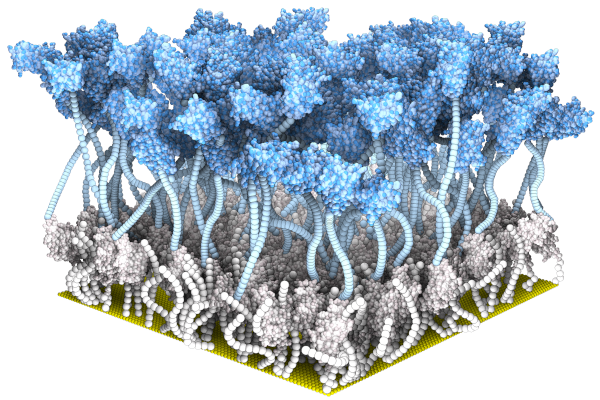
*Guo K, *Wustoni S, *Koklu A, Diaz-Galicia E, Moser M, Hama A, Alqahtani
AA, Ahmad AN, Alhamlan FS, Shuaib M, Pain A, McCulloch
I, #Arold ST, #Grünberg
R, #Inal S (2021) Rapid
Single-Molecule Detection of COVID-19 and MERS Antigens via
Nanobody-Functionalized Organic Electrochemical Transistors. Nature Biomedical
Engineering
Guo K et al. #Arold ST, #Grünberg R, #Inal S (2023) SpyDirect enables autocatalytic and high-density nanobody functionalization for robust protein sensing. (submitted) (* shared first co-authors; # shared corresponding) additional papers in Advanced Materials and others, plus several patents |
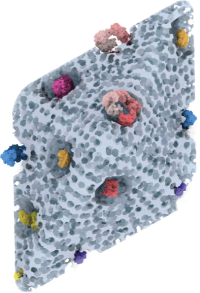
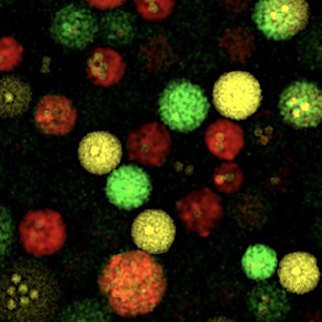
| Therapeutic multi-enzyme Nanoreactors |
| in collaboration with Niveen Kashab and her Smart Hybrid Materials Lab as well as with Jasmmeen Merzaban and her team. |
|
|
|
*Sharip A, *Qutub SS, Baslyman W,
Zhao L, #Khashab
NM, #Arold
ST, #Grünberg R
(2023) Hierarchically engineered multi-enzyme nanoreactors for in vitro
and intracellular drug biosynthesis. (submitted)
(* shared first co-authors; # shared corresponding) |
| Community | |

|
Synthetic Biology Open Language (SBOL) Founding member, Editor (2015 - 2017) |

|
iGEM Team advisor to the Freiburg-Software team (2009) and the Seattle robotics "team" (2009) Chair of the iGEM software track (2014 - 2016) |

|
EMBO Global Exchange "Introduction to Synthetic Biology" (Buenos
Aires, 2012) Co-organizer. The course and related activities helped kick-starting synbio in Latin America. |
| Lab automation |
|
|
*Ramos-Mandujano G, *Grünberg R et al., Li M (2023) An Open-Source, Automated, and Cost-Effective Platform for COVID-19 Diagnosis and Rapid Portable Genomic Surveillance Using Nanopore Sequencing. (submitted) [GitHub] (* shared first co-authors) |

| Software Development |
|

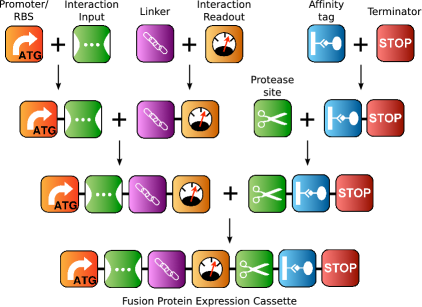

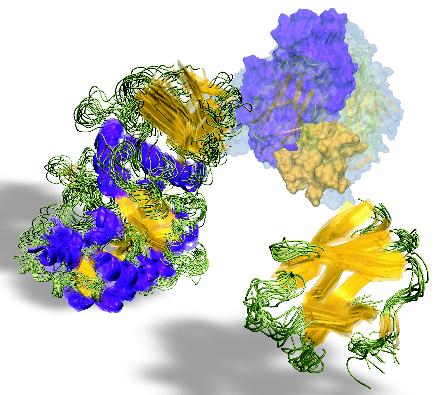
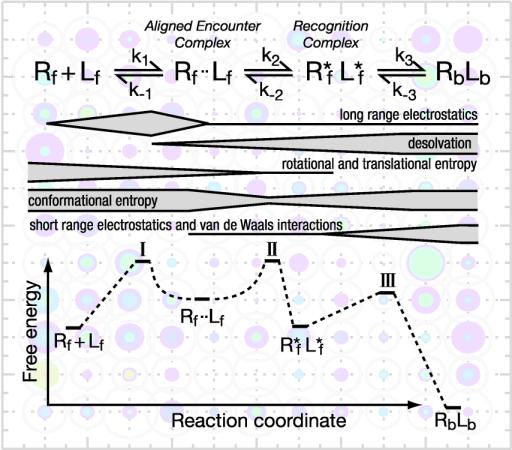
![[view]](img/evo_poster_hd.jpg){kind=link}