|
Initially, docking studies (combined with experimental examinations) have been employed to assist the search for an optimal substrate mimetic. Since the results obtained proved to be useful for explaining the enzymes new specifities, the studies were later extended to model peptides containing Arg, Lys, Tyr or Phe. An example of the latter results is presented here.
|
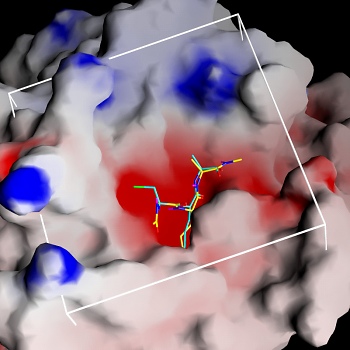
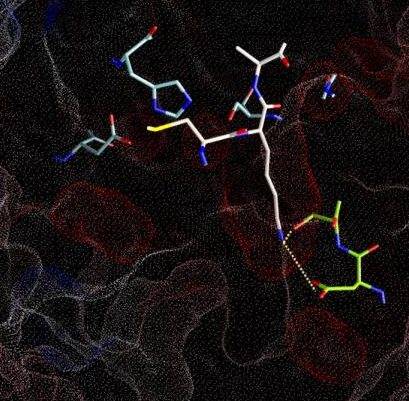
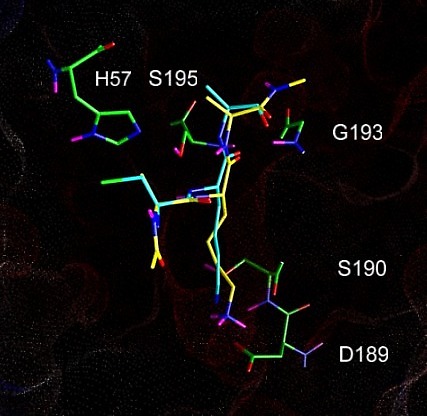
Figure 4.2a shows the docked arrangement of Ac-Ala-Lys-Ala-Me bound to wt-trypsin. The lowest energy structure found (yellow) resembles the arrangement of SBTI's central residues Cys-Lys-Ala (blue, the same as shown in Fig. 1a) in the pdb structure 3tgi with an RMSD of only 0,66 A˛. This result is a good confirmation of the accuratness of docking studies and has encouraged further studies with this system.
The box embedded into the protein describes the volume (22 x 22 x 22 A) searched during the docking simulation (i.e. the dimension of the energy grid employed).
Click on the image for a more detailed view.
|
|
Fig. 4.2a: Docking of a Lys-containing peptide (AKA, yellow) to wt-trypsin. The "real" arrangment, as observed in the x-ray structure 3tgi, is given in blue. |
|
Experiments revealed a (weak) specifity of trypsin D189A,S190A towards Tyr-containing substrates (see 4.1). As discussed above, this contradicted the assumption that Oy of S190 facilitates Tyr-binding by electrostatic interactions with the Tyr side chain.
The binding of the peptide Ala-Tyr-Ala to trypsin D189A,S190A was examined in docking studies. The results offer an explanation for this effect and are a good example of how to use docking simulations for connecting experimental results with structural considerations.
|
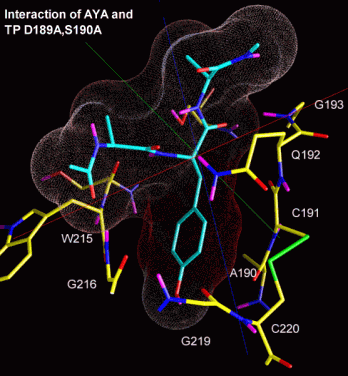
The docked arrangement of Ac-Ala-Tyr-Ala-Me bound to trypsin D189A,S190A is given in figure 4.2b. The ligand's position is very similar to the one observed in the docking against wt-trypsin (not shown).
The lowest energy structure found is consistent to the catalytic mechanism, with the carbonyl carbon situated next to the attacking S195 and the carbonyl oxygen positioned in the oxanion whole (S195 and G193). Clearly, the Tyr sidechain is not pointing to A190. Its OH-group, by contrast, seems to interact with the backbone oxygens both of G219 and (less obvious) A190.
The ligand surface is colored in accordance to the local interaction energy. The most evident interaction is calculated for the Tyr side chain's aromatic carbons.
|
|
Fig. 4.2b: Docking of a Tyr-containing peptide (blue) to trypsin D189A,S190A. Projected onto the van-der-Waals surface is each ligand atom's contribution to the interaction (red: neg. interaction energy, i.e. attraction, blue: positive interaction energy, i.e. repulsion). Protein residues interacting with the ligand are drawn in yellow. |
|
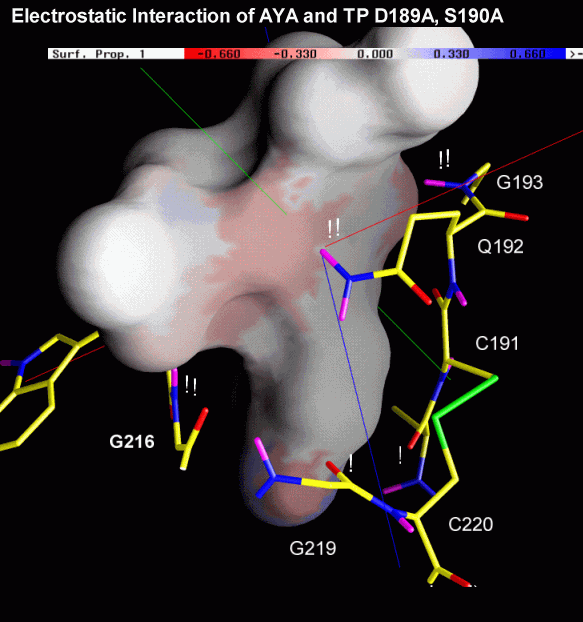
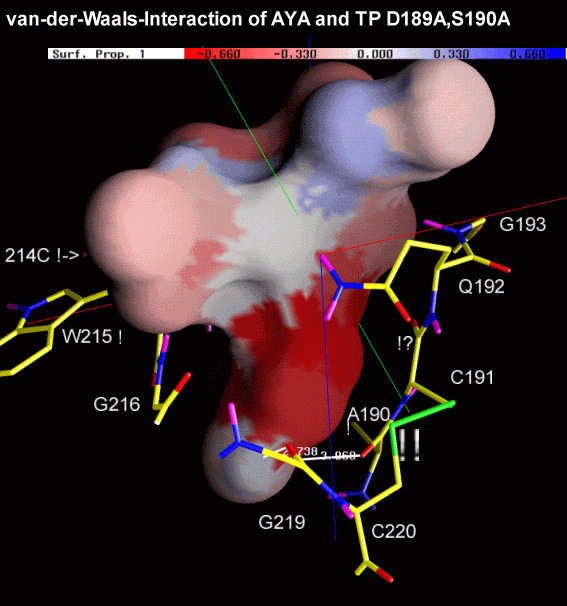
The next two figures are based on the same docking result. This time, the ligand is rendered with a solid surface. Again, the surface is colored according to the underlying ligand atom's interaction energy. In contrast to figure 4.2.b however, electrostatic and van-der-Waals interaction terms are displayed seperately.
Click the images to view an enlarged version.
|
Fig. 4.2c: Electrostatic (left) and van-der-Waals (right) interaction of AYA and Trypsin D189A,S190A. The surface color follows the local interaction energy (red: negative, blue: positive). Selected protein carbons are drawn yellow, atoms especially contributing to the interaction are marked with "!". |
Considering the above figure, the experimental findings can be explained: Binding of the Tyr side chain is not adversely affected by the mutation S190A, because the hydroxy group of Tyr can electrostatically interact with backbone oxygens of G219 and A(S)190. On the contrary, removing Oy of S190, in fact, promotes binding since this atom apparently "obstructs" van-der-Waals interaction between S190's Cß and aromatic ligand atoms.
As highlighted by the right figure, overall binding is mainly dominated by relatively unspecific van-der-Waals attraction between ligand atoms and the protein backbone.
|
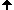
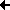
 webmaster
webmaster